SpatialArtifacts Tutorial
Harriet Jiali He
Johns Hopkins University, Baltimore, MD, United StatesStephanie C. Hicks
Johns Hopkins University, Baltimore, MD, United States2026-05-04
Source:vignettes/hippocampus-edge-detection.Rmd
hippocampus-edge-detection.RmdIntroduction
SpatialArtifacts is an R package that provides a
data-driven a two-step workflow to identify, classify, and handle
spatial artifacts in spatial transcriptomics data from multiple
platforms including 10x Visium (Standard and HD).
Broadly, the idea behind the package is we that combine median absolute
deviation (MAD)-based outlier detection with morphological image
processing to identify the artifacts. These artifacts, often appearing
as areas of low gene/UMI counts or high mitochondrial ratio at tissue
edges (edge artifacts) or in the interior
(interior artifacts), can negatively impact downstream
analyses. The methods are implemented as an R package within the
Bioconductor framework, and is available via SpatialArtifacts.
In the following, we provide an overview of the functionality in the package and we demonstrate how to apply the package on real-world datasets across different spatial transcriptomics platforms.
More details describing the method will be available in our upcoming preprint.
Installation
The latest development version can also be installed from the
devel version of Bioconductor:
BiocManager::install("SpatialArtifacts", version="devel")
install.packages("BiocManager")
BiocManager::install("SpatialArtifacts")The latest development version can also be installed from the
devel version of Bioconductor or from GitHub.
Input data format
In the examples below, we assume the input data are provided as a
SpatialExperiment
Bioconductor object. In this case, the outputs are stored in the
rowData of the SpatialExperiment object.
Platform Support
SpatialArtifacts is designed to work across multiple
spatial transcriptomics platforms:
- Standard Visium (55µm bins, hexagonal grid): ~5,000 spots per capture area
- VisiumHD 16µm (16µm bins, square grid): ~480,000 bins per capture
area
- VisiumHD 8µm (8µm bins, square grid): ~1,920,000 bins per capture area
IMPORTANT: The morphological detection framework automatically adapts to different grid arrangements, but parameter scaling is critical for optimal performance across platforms.
Two key steps
The core philosophy is a two-step process: detect, and then classify. This separates the sensitive task of identifying all potential problem spots from the more nuanced task of deciding what to do with them.
The detection phase
In the first step, we use the detectEdgeArtifacts()
function. Here, the goal is to identify all spots that could potentially
be part of an artifact.
How does it work:
-
Outlier identification: Find spots with abnormally
low QC metrics (e.g.,
sum_gene) using a Median Absolute Deviation (MAD) threshold.
-
Morphological cleaning: Apply sequential
raster-based focal operations through
focal_transformations():-
3×3 fill (
my_fill): Fill spots completely surrounded by outliers -
5×5 outline (
my_outline): Fill spots outlined by outliers in a larger 16-pixel perimeter -
Star pattern (
my_fill_star): Fill spots with outliers in all four cardinal directions (N, S, E, W) -
Small cluster removal: Remove isolated normal
regions below
min_cluster_sizethreshold (default: 40 spots)
- Use 8-directional connectivity for connected component analysis
-
3×3 fill (
-
Cluster detection: Group these outliers into
contiguous “problem areas” (
problemAreas) -
Edge identification: Evaluate whether clusters
touch tissue boundaries using
clumpEdges(). For each of the four borders (north, south, east, west), calculates the proportion of boundary spots belonging to each cluster. A cluster is classified as an edge artifact if this proportion meets or exceedsedge_threshold(default: 0.75, meaning ≥75% border coverage) on any single border direction
The output from this phase will add three raw columns to
your spe object: _edge,
_problem_id, and _problem_size
The classification phase
In the second step, we use the classifyEdgeArtifacts()
function. Here the goal is to take the raw detections from the previous
step and apply a clear, hierarchical logic to assign final labels.
How does it work:
-
Input: Requires the
speobject processed bydetectEdgeArtifacts()
-
Hierarchical Classification: Apply a 2×2 logic
system based on Location and Size:
-
Location: Is the artifact an
_edge_artifact(TRUEorFALSE) based on the detection step?
-
Size: Is the artifact’s
_problem_sizelarger thanmin_spots(default:20)?
-
Location: Is the artifact an
-
Label Assignment: This logic produces five
intuitive categories:
-
"not_artifact"— High-quality spots
-
"large_edge_artifact"— Large artifact cluster (> min_spots) touching the tissue edge
-
"small_edge_artifact"— Small artifact cluster (≤ min_spots) touching the tissue edge
-
"large_interior_artifact"— Large artifact cluster (> min_spots) located inside the tissue
-
"small_interior_artifact"— Small artifact cluster (≤ min_spots) located inside the tissue
-
The output from this phase will add one classification column named
_classification.
Helpful information on parameters
Tuning the parameters lets you adapt the workflow to different tissue types, data quality, and spatial transcriptomics platforms. The package uses a wrapper function that routes to platform-specific implementations.
Platform selection
CRITICAL FIRST STEP: Specify your platform using the
platform parameter in detectEdgeArtifacts()
function:
| Platform | Function Call | Required Parameters |
|---|---|---|
| Standard Visium | detectEdgeArtifacts(spe, platform="visium", ...) |
(none required) |
| VisiumHD | detectEdgeArtifacts(spe, platform="visiumhd", resolution="16um", ...) |
resolution (“8um” or “16um”) |
Example use cases
# Standard Visium (55µm hexagonal grid)
spe <- detectEdgeArtifacts(spe, platform = "visium", ...)
# VisiumHD 16µm (square grid)
spe <- detectEdgeArtifacts(spe, platform = "visiumhd", resolution = "16um", ...)
# VisiumHD 8µm (square grid)
spe <- detectEdgeArtifacts(spe, platform = "visiumhd", resolution = "8um", ...)Parameters for detectEdgeArtifacts()
The wrapper function accepts platform-specific parameters that are routed to the appropriate implementation.
For all platforms
-
platform(REQUIRED) – Character string:"visium"or"visiumhd"(case insensitive)- Determines which platform-specific function to use
- No default value; must be explicitly specified for clarity
-
qc_metric(Default:"sum_gene") – Column name for QC metric used in outlier detection- Common alternatives:
"sum_umi","detected","nFeature" - The function will auto-detect some common variants
- Common alternatives:
-
samples(Default:"sample_id") – Column name for sample identifiers- Each sample is processed independently
-
mad_threshold(Default: 3) – Sensitivity for detecting outliers- Lower values (1.5–2) are more sensitive
- Higher values (3–4) are more conservative
-
name(Default:"edge_artifact") – Prefix for output column names- Outputs:
[name]_edge,[name]_problem_id,[name]_problem_size
- Outputs:
verbose(Default:TRUE) – Print progress messageskeep_intermediate(Default:FALSE) – Retain intermediate outlier detection columns
For standard Visium
When platform = "visium", use:
-
edge_threshold(Default: 0.75) – Minimum proportion of a tissue boundary that must be occupied by outlier clusters (collectively) for those clusters to be classified as edge artifacts.Important Behavior: The threshold is applied to the total coverage of all outlier clusters on each boundary direction (North, South, East, West). If multiple clusters collectively cover ≥
edge_thresholdof a boundary, all clusters touching that boundary are classified as edge artifacts, even if no single cluster meets the threshold individually.Example Scenario:
- North boundary contains 100 spots total
- Cluster A occupies 30 boundary spots (30% coverage)
- Cluster B occupies 50 boundary spots (50% coverage)
- Combined coverage: 80% ≥ 75% threshold
- Result: Both Cluster A and Cluster B are classified as edge artifacts
Rationale: Edge drying artifacts typically affect large, continuous regions along tissue boundaries. Multiple clusters on the same boundary often result from a single underlying technical failure (incomplete permeabilization) and should be treated as a unified artifact rather than independent events.
Tuning Guidance:
- Higher values (0.75–0.90): More conservative, captures only large-scale boundary failures
- Lower values (0.40–0.60): More sensitive, may flag smaller boundary-adjacent regions
- Very low values (<0.30): Aggressive, may misclassify biological low-expression zones as artifacts
-
min_cluster_size(Default: 40) – Minimum cluster size (in spots) for morphological cleaning during focal transformation steps- Isolated “normal” regions smaller than this threshold within outlier areas will be filled in to create contiguous artifact regions
- For Standard Visium: 40 spots ≈ 0.12 mm² physical area
-
shifted(Default:FALSE) – Apply coordinate adjustment for hexagonal grid alignment- Keep the default FALSE when using array_row/array_col coordinates from Space Ranger, as rasterFromXYZ handles the hexagonal grid spacing automatically.
- Only set to TRUE if using a custom coordinate system where odd columns require manual offset correction.
-
batch_var(Default:"both") – Determines grouping for MAD calculation- Options:
"sample_id","slide", or"both" -
"both": Spots flagged as outliers if below threshold in either sample or slide grouping
- Options:
For VisiumHD
When platform = "visiumhd", use:
-
resolution(REQUIRED) – Character string:"8um"or"16um"- Specifies the bin size of your VisiumHD data
- This is mandatory for VisiumHD; the function will error if not provided
- Determines conversion from physical units (µm) to bins
-
buffer_width_um(Default: 80) – Buffer zone width in micrometers (physical units)- Defines the edge region where artifacts are expected
- Automatically converted to bins based on
resolution:- At 16µm resolution: 80 µm → 5 bins
- At 8µm resolution: 80 µm → 10 bins
-
Tuning guidance:
- Increase (100-150 µm) for tissues with larger edge artifacts
- Decrease (50-60 µm) for precise edge detection
-
min_cluster_area_um2(Default: 1280) – Minimum cluster area in square micrometers (physical units)- Clusters smaller than this will be filtered out during morphological cleaning
- Automatically converted to bins based on
resolution:- At 16µm resolution: 1280 µm² → 5 bins (16×16 µm per bin)
- At 8µm resolution: 1280 µm² → 20 bins (8×8 µm per bin)
- Physical consistency: Same area threshold gives different bin counts at different resolutions
- Default (1280 µm²) was calibrated for 16µm resolution
-
col_xandcol_y(Default:"array_col","array_row") – Column names for bin coordinates- Important: These should be bin indices, not pixel coordinates
- Using bin indices is much more memory-efficient than pixel coordinates
Key Difference from Visium: VisiumHD parameters are specified in physical units (µm, µm²) rather than bin counts. This ensures consistency across resolutions while the algorithm handles the bin conversion internally.
Parameters for classifyEdgeArtifacts()
The classification step is platform-independent but requires appropriate parameter scaling.
-
min_spots(Default: 20) – CRITICAL PARAMETER: Threshold (in number of spots/bins) to distinguish"large"from"small"artifactsPlatform-Specific Scaling Required:
This parameter must be adjusted based on spatial resolution to represent equivalent physical artifact sizes:
Platform Recommended min_spotsPhysical Area Scaling Factor Standard Visium (55µm) 20-40~0.06-0.12 mm² Baseline (1×) VisiumHD 16µm bins 100-200~0.026-0.051 mm² ~6-10× Visium VisiumHD 8µm bins 400-800~0.026-0.051 mm² ~20-40× Visium Automatic Scaling Formula:
min_spots_HD <- min_spots_visium × (55 / bin_size_µm)² # Example: For min_spots = 30 on Standard Visium # VisiumHD 16µm: 30 × (55/16)² ≈ 354 bins # VisiumHD 8µm: 30 × (55/8)² ≈ 1,420 binsWhy scaling matters: The same physical artifact (e.g., 0.1 mm² edge dryspot) will cover:
- Standard Visium: ~33 spots
- VisiumHD 16µm: ~390 bins
- VisiumHD 8µm: ~1,560 bins
Without scaling, large VisiumHD artifacts would be incorrectly classified as “small.”
qc_metric(Default:"sum_umi") – QC metric column for validation (must exist but not directly used in classification logic)samples(Default:"sample_id") – Sample ID column name-
exclude_slides(Default:NULL) – Vector of slide IDs to exclude from edge classification- Spots on these slides will have edge artifact status set to
FALSE
- Spots on these slides will have edge artifact status set to
name(Default:"edge_artifact") – Must match the name used indetectEdgeArtifacts()
Platform Comparison Summary
| Feature | Standard Visium | VisiumHD |
|---|---|---|
| Grid Type | Hexagonal | Square |
Requires shifted? |
No (default FALSE) | No (not used) |
| Resolution Parameter | Not applicable |
Required ("8um" or
"16um") |
| Edge Detection Method | Morphological + boundary coverage | Buffer zone + morphological |
| Parameter Units | Spot counts | Physical units (µm, µm²) |
Default min_spots (classify) |
20-40 | 100-200 (16µm), 400-800 (8µm) |
| Typical Dataset Size | ~5,000 spots | ~480k bins (16µm), ~1.9M bins (8µm) |
Understanding the output columns
After both functions, several columns are added to
colData(spe):
-
*_edge– Raw detection: Is the spot in a cluster touching the tissue border? (TRUE/FALSE)
-
*_problem_id– Raw detection: ID of the problem area.
-
*_problem_size– Raw detection: Size (number of spots) of the problem area.
-
*_true_edges— Intermediate: Edge status after applyingexclude_slides(used byclassifyEdgeArtifacts()). -
*_classification— Final classification: One of"not_artifact","large_edge_artifact","small_edge_artifact","large_interior_artifact", or"small_interior_artifact".
Example: Standard Visium workflow
This package includes spe_vignette, a lightweight
SpatialExperiment object derived from a human hippocampus
Visium sample.
This vignette will load this raw-like object and run the full
SpatialArtifacts workflow on it live.
Note: To meet package size requirements (< 5MB), this object has been subset (e.g., to coding genes) and sparsified, but no artifact detection has been run. We will perform those steps now.
Data preparation: converting to dense matrix
The underlying spatial clustering functions in this package currently
require a dense matrix to perform coordinate-based
calculations. We must first convert the sparse counts assay
in our spe_vignette object to a standard (dense)
matrix.
data(spe_vignette)
# Loaded data dimensions:
dim(spe_vignette)
#> [1] 12971 4965
assay(spe_vignette, "counts") <- as.matrix(assay(spe_vignette, "counts"))
names(colData(spe_vignette))[names(colData(spe_vignette)) == "sum"] <- "sum_umi"
spe_detected <- detectEdgeArtifacts(
spe_vignette,
platform = "visium", # IMPORTANT: Specify Standard Visium platform
qc_metric = "sum_umi",
samples = "sample_id",
batch_var = "sample_id",
mad_threshold = 3,
edge_threshold = 0.75,
name = "edge_artifact"
)
#> Detecting edges...
#> Sample V11L05-335_C1: 74 edge spots detected
#> Finding problem areas...
#> Removed intermediate columns: lg10_sum_umi, sum_umi_3MAD_outlier_sample, sum_umi_3MAD_outlier_binary
#> Edge artifact detection completed!
#> Total edge spots: 74
#> Total problem area spots: 78
# === RESULTS ===
table(Edge_Detected = spe_detected$edge_artifact_edge)
#> Edge_Detected
#> FALSE TRUE
#> 4891 74
# Classification with Standard Visium parameters
spe_classified <- classifyEdgeArtifacts(
spe_detected,
min_spots = 20,
name = "edge_artifact"
)
#> Classifying artifacts spots...
#> Classification added: edge_artifact_classification
#>
#> Classification summary:
#> not_artifact: 4887 spots
#> small_edge_artifact: 74 spots
#> small_interior_artifact: 4 spots
# === Classification Results ===
table(spe_classified$edge_artifact_classification)
#>
#> not_artifact small_edge_artifact small_interior_artifact
#> 4887 74 4VisiumHD Workflow Example
For VisiumHD data, the workflow is identical except for parameter scaling. Here’s a complete example showing how to adapt parameters for VisiumHD:
VisiumHD 16µm Resolution Example
# This is a pseudo-example demonstrating VisiumHD 16µm workflow
# Assumes you have loaded a VisiumHD SpatialExperiment object as 'spe_hd16'
# Step 1: Ensure required QC metrics are calculated
library(scuttle)
spe_hd16 <- addPerCellQCMetrics(spe_hd16)
# Step 2: Detection Phase - VisiumHD uses square grid (no 'shifted' needed)
spe_hd16_detected <- detectEdgeArtifacts(
spe_hd16,
platform = "visiumhd", # Specify VisiumHD platform
resolution = "16um", # REQUIRED for VisiumHD
qc_metric = "sum_umi", # or "sum" depending on your colData
samples = "sample_id",
buffer_width_um = 100, # VisiumHD-specific parameter
mad_threshold = 2.5,
edge_threshold = 0.75,
name = "edge_artifact"
)
# Step 3: Classification Phase - CRITICAL: Scale min_spots for VisiumHD resolution
# For 16µm bins, use ~6-10× the Standard Visium threshold
min_spots_16um <- 30 * (55 / 16)^2 # ≈ 354 bins
spe_hd16_classified <- classifyEdgeArtifacts(
spe_hd16_detected,
qc_metric = "sum_umi",
min_spots = round(min_spots_16um), # ~350 bins
name = "edge_artifact"
)
# Visualization (same approach as Standard Visium)
table(spe_hd16_classified$edge_artifact_classification)VisiumHD 8µm Resolution Example
# This is a pseudo-example demonstrating VisiumHD 8µm workflow
# Assumes you have loaded a VisiumHD 8µm SpatialExperiment object as 'spe_hd8'
# Step 1: QC metrics
spe_hd8 <- addPerCellQCMetrics(spe_hd8)
# Step 2: Detection Phase
spe_hd8_detected <- detectEdgeArtifacts(
spe_hd8,
platform = "visiumhd", # Specify VisiumHD platform
resolution = "8um", # REQUIRED: Specify 8µm resolution
qc_metric = "sum_umi",
samples = "sample_id",
buffer_width_um = 100, # Buffer zone in micrometers
mad_threshold = 2.5,
edge_threshold = 0.75,
name = "edge_artifact"
)
# Step 3: Classification with 8µm-appropriate threshold
# For 8µm bins, use ~20-40× the Standard Visium threshold
min_spots_8um <- 30 * (55 / 8)^2 # ≈ 1,420 bins
spe_hd8_classified <- classifyEdgeArtifacts(
spe_hd8_detected,
qc_metric = "sum_umi",
min_spots = round(min_spots_8um), # ~1,400 bins
name = "edge_artifact"
)
table(spe_hd8_classified$edge_artifact_classification)Key VisiumHD Considerations
Platform-Specific Function Calls:
| Platform | Function Call | Required Parameters |
|---|---|---|
| Standard Visium | detectEdgeArtifacts(..., platform="visium") |
(none required) |
| VisiumHD 16µm | detectEdgeArtifacts(..., platform="visiumhd", resolution="16um") |
resolution |
| VisiumHD 8µm | detectEdgeArtifacts(..., platform="visiumhd", resolution="8um") |
resolution |
Parameter Recommendations by Platform:
| Parameter | Standard Visium | VisiumHD 16µm | VisiumHD 8µm |
|---|---|---|---|
platform |
"visium" |
"visiumhd" |
"visiumhd" |
resolution |
N/A (not used) | "16um" |
"8um" |
shifted |
FALSE (default) | N/A (handled internally) | N/A |
buffer_width_um |
N/A |
100 (default) |
100 (default) |
mad_threshold |
1.5-3.0 | 2.0-3.0 | 2.0-3.0 |
min_spots (classify) |
20-40 | 100-200 | 400-800 |
| Grid Type | Hexagonal | Square | Square |
Visualization: QC Metrics and Detection Results
We’ll create a comprehensive visualization showing QC metrics, detection results, and detailed cluster information:
library(SpatialExperiment)
library(patchwork)
plot_data <- as.data.frame(colData(spe_classified))
plot_data <- cbind(plot_data, as.data.frame(spatialCoords(spe_classified)))
plot_data_in_tissue <- plot_data[plot_data$in_tissue, ]
base_theme <- theme_void() +
theme(plot.title = element_text(size = 12, hjust = 0.5), legend.position = "right")
.plt <- \(df, col, fun = \(.) .) {
ggplot(df, aes(x = pxl_col_in_fullres, y = pxl_row_in_fullres, col = fun(.data[[col]]))) +
geom_point(size = 0.5) +
base_theme +
coord_fixed()
}
plot_data_in_tissue$raw_problem <- !is.na(plot_data_in_tissue$edge_artifact_problem_id)
plot_data_in_tissue$cluster_display <- plot_data_in_tissue$edge_artifact_problem_id
plot_data_in_tissue$artifact_type <- "Normal"
plot_data_in_tissue$artifact_type[plot_data_in_tissue$edge_artifact_edge] <- "Edge Artifact"
plot_data_in_tissue$artifact_type[!is.na(plot_data_in_tissue$edge_artifact_problem_id) & !plot_data_in_tissue$edge_artifact_edge] <- "Interior Problem"
p1 <- .plt(plot_data_in_tissue, "sum_umi", \(.) log10(.+1)) +
scale_color_viridis_c(name = "log10(UMI)") + ggtitle("A. UMI Counts")
p2 <- .plt(plot_data_in_tissue, "detected") +
scale_color_viridis_c(name = "Genes", option = "plasma") + ggtitle("B. Detected Genes")
p3 <- .plt(plot_data_in_tissue, "raw_problem") +
scale_color_manual(values = c("FALSE" = "lightgray", "TRUE" = "red"), name = "Problem?") + ggtitle("C. Raw Detection")
p4 <- .plt(plot_data_in_tissue, "cluster_display") +
scale_color_discrete(name = "Cluster ID", na.value = "lightgray") + ggtitle("D. Problem Area Clusters") + theme(legend.key.size = unit(0.3, "cm"), legend.text = element_text(size = 8))
p5 <- .plt(plot_data_in_tissue, "artifact_type") +
scale_color_manual(values = c("Normal" = "lightgray", "Edge Artifact" = "red", "Interior Problem" = "blue"), name = "Type") + ggtitle("E. Edge vs Interior")
p6 <- .plt(plot_data_in_tissue, "edge_artifact_classification") +
scale_color_manual(values = c("not_artifact" = "lightgray", "large_edge_artifact" = "red", "small_edge_artifact" = "orange", "large_interior_artifact" = "blue", "small_interior_artifact" = "cyan"), name = "Final Class", na.value = "grey50") + ggtitle("F. Hierarchical Classification")
(p1 | p2) / (p3 | p4) / (p5 | p6)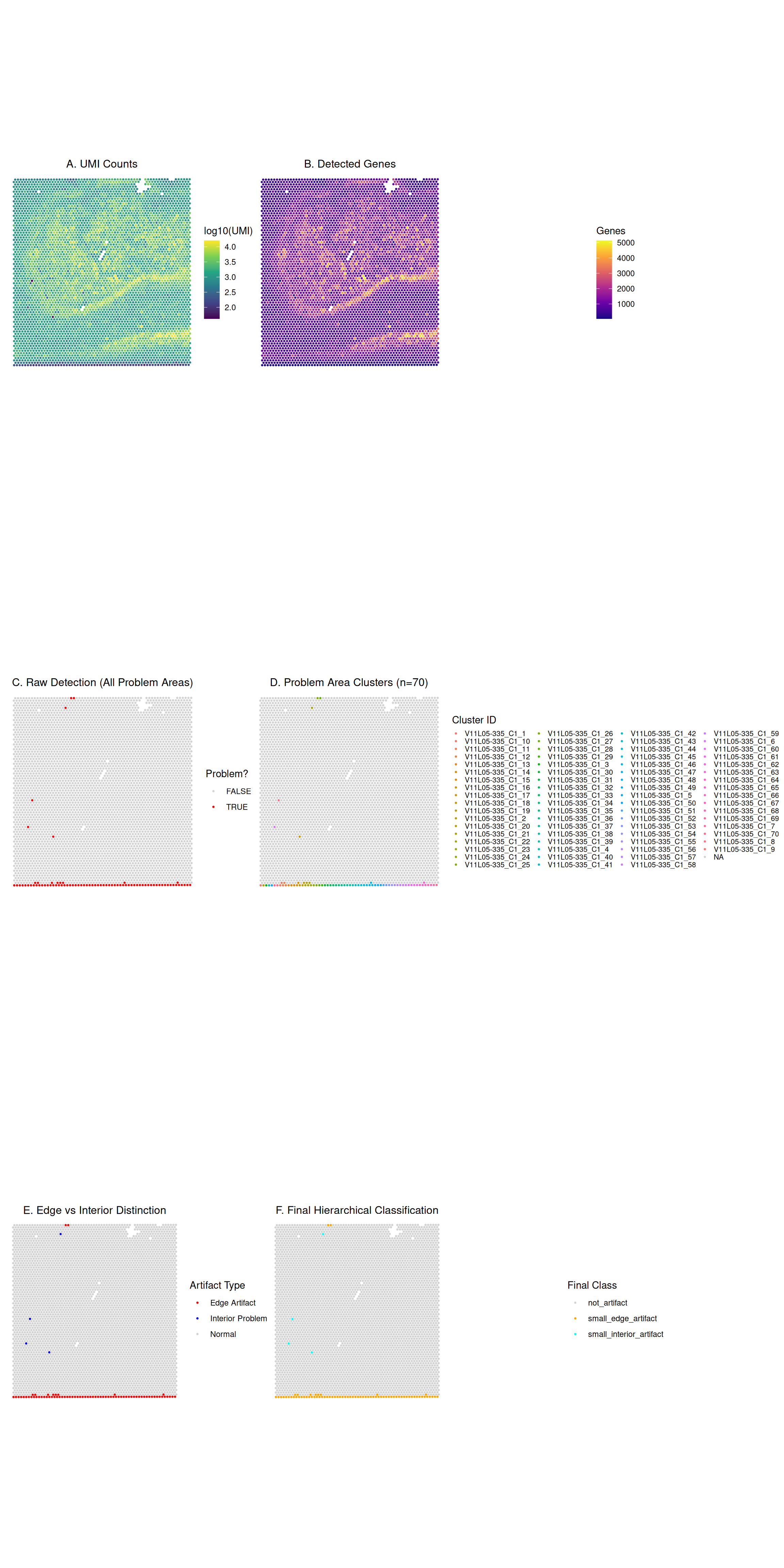
Classification Summary
Let’s examine the enhanced classification system:
Quality Control Validation
Finally, let’s validate that flagged spots have lower quality metrics:
| Metric | Flagged (Edge) | Non-flagged | Difference |
|---|---|---|---|
| Median UMI | 120 | 1784 | 1664 |
| Median Detected Genes | 106 | 1019 | 912 |
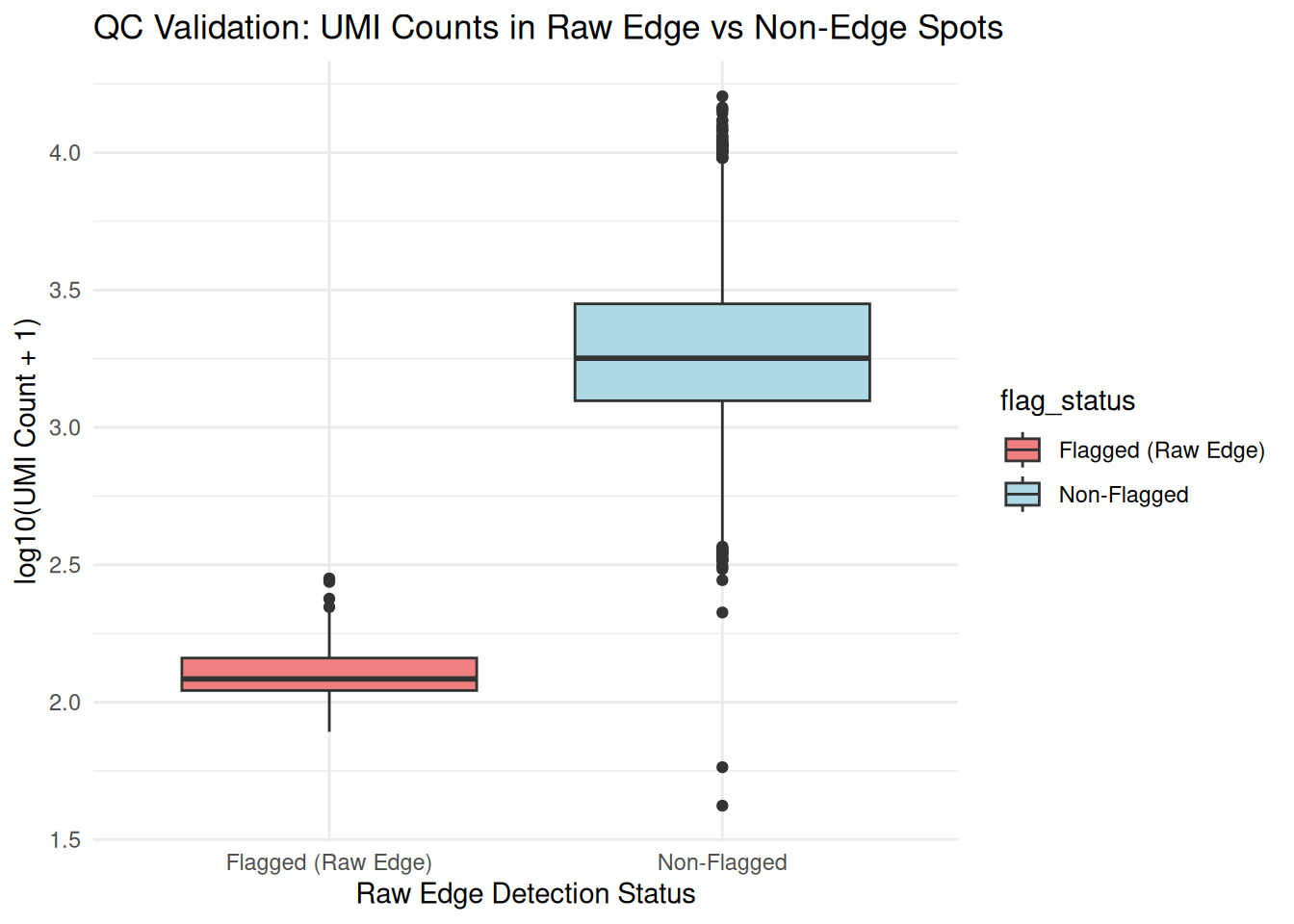
Filtering Out Problematic Spots (Optional)
Based on the new classifications, users can make informed decisions about filtering. For example, you might decide to remove all edge artifacts (both large and small) while keeping interior artifacts for further review.
Here’s how you can filter the SpatialExperiment object
to remove all spots classified as both
"large_edge_artifact" or
"small_edge_artifact":
if ("edge_artifact_classification" %in% names(colData(spe_classified))) {
spots_to_keep <- !spe_classified$edge_artifact_classification %in%
c("large_edge_artifact", "small_edge_artifact")
spe_filtered <- spe_classified[, spots_to_keep]
message("Original number of spots: ", ncol(spe_classified))
message("Number of spots after filtering: ", ncol(spe_filtered))
} else {
message("Classification column not found. Filtering step skipped.")
}
#> Original number of spots: 4965
#> Number of spots after filtering: 4891
plot_data_before <- as.data.frame(colData(spe_classified))
plot_data_before <- cbind(plot_data_before, as.data.frame(spatialCoords(spe_classified)))
plot_data_before_in_tissue <- plot_data_before[plot_data_before$in_tissue, ]
plot_data_after <- as.data.frame(colData(spe_filtered))
if (ncol(spe_filtered) > 0) {
plot_data_after <- cbind(plot_data_after, as.data.frame(spatialCoords(spe_filtered)))
}
p1_umi_before <- .plt(plot_data_before_in_tissue, "sum_umi", \(.) log10(.+1)) +
scale_color_viridis_c(name = "log10(UMI)") + ggtitle("UMI Counts (Before Filtering)")
p3_class_before <- .plt(plot_data_before_in_tissue, "edge_artifact_classification") +
scale_color_manual(values = c("not_artifact" = "lightgray", "large_edge_artifact" = "red", "small_edge_artifact" = "orange", "large_interior_artifact" = "blue", "small_interior_artifact" = "cyan"), name = "Final Class", na.value = "grey50", drop = FALSE) + ggtitle("Classification (Before Filtering)")
if (ncol(spe_filtered) > 0) {
p2_umi_after <- .plt(plot_data_after, "sum_umi", \(.) log10(.+1)) +
scale_color_viridis_c(name = "log10(UMI)") + ggtitle("UMI Counts (After Filtering)")
p4_class_after <- .plt(plot_data_after, "edge_artifact_classification") +
scale_color_manual(values = c("not_artifact" = "lightgray", "large_edge_artifact" = "red", "small_edge_artifact" = "orange", "large_interior_artifact" = "blue", "small_interior_artifact" = "cyan"), name = "Final Class", na.value = "grey50", drop = FALSE) + ggtitle("Classification (After Filtering)")
} else {
p2_umi_after <- ggplot() + theme_void() + ggtitle("UMI Counts (After Filtering - No Spots)")
p4_class_after <- ggplot() + theme_void() + ggtitle("Classification (After Filtering - No Spots)")
}
combined_filtering_plot_2x2 <- (p1_umi_before | p2_umi_after) / (p3_class_before | p4_class_after)
print(combined_filtering_plot_2x2)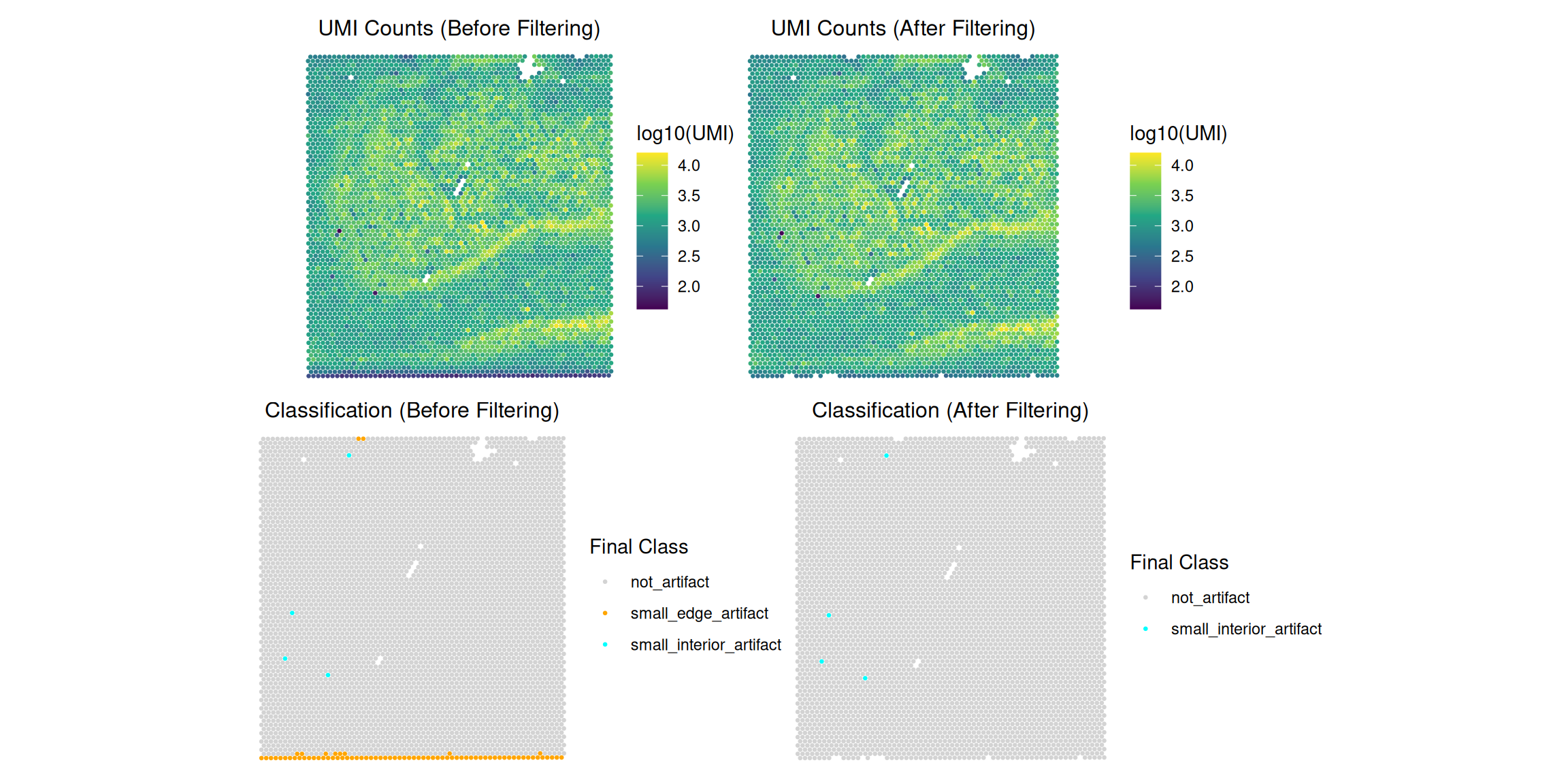
Conclusion
This vignette demonstrated the SpatialArtifacts workflow across multiple spatial transcriptomics platforms. Specifically, it showed:
- Standard Visium workflow: Complete example using the included hippocampus dataset
-
VisiumHD adaptation: How to properly scale
parameters for 16µm and 8µm resolution data
- Platform-specific considerations: Grid arrangement, parameter scaling formulas, and typical artifact sizes
-
Visualization: Display hierarchical classification
results alongside QC metrics
- Classification logic: Distinguish between edge vs. interior and large vs. small artifacts
- Validation: Flagged spots show significantly lower molecular capture
Key Takeaways for Multi-Platform Usage:
Grid Structure: Standard Visium uses hexagonal grids (shifted = FALSE by default), while VisiumHD uses square grids (shifted parameter not used)
-
Critical Parameter Scaling: The
min_spotsthreshold inclassifyEdgeArtifacts()must scale with spatial resolution:- Use the formula:
min_spots_HD = min_spots_visium × (55 / bin_size)² - Without scaling, artifacts will be incorrectly classified by size
- Use the formula:
Morphological Framework: The same detection logic works across platforms, automatically adapting to different grid arrangements
Physical Consistency: Scaled parameters ensure that “large” and “small” artifact classifications represent equivalent physical sizes regardless of platform
Overall, SpatialArtifacts provides a unified, platform-agnostic framework for detecting and classifying spatial artifacts, enabling consistent quality control across the evolving spatial transcriptomics technology landscape.
Session Information
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] dplyr_1.2.1 patchwork_1.3.2
#> [3] ggplot2_4.0.3 SpatialArtifacts_1.1.0
#> [5] SpatialExperiment_1.22.0 SingleCellExperiment_1.34.0
#> [7] SummarizedExperiment_1.42.0 Biobase_2.72.0
#> [9] GenomicRanges_1.64.0 Seqinfo_1.2.0
#> [11] IRanges_2.46.0 S4Vectors_0.50.0
#> [13] BiocGenerics_0.58.0 generics_0.1.4
#> [15] MatrixGenerics_1.24.0 matrixStats_1.5.0
#> [17] BiocStyle_2.40.0
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 rjson_0.2.23 xfun_0.57
#> [4] bslib_0.10.0 lattice_0.22-9 vctrs_0.7.3
#> [7] tools_4.6.0 parallel_4.6.0 tibble_3.3.1
#> [10] pkgconfig_2.0.3 Matrix_1.7-5 RColorBrewer_1.1-3
#> [13] S7_0.2.2 desc_1.4.3 lifecycle_1.0.5
#> [16] compiler_4.6.0 farver_2.1.2 textshaping_1.0.5
#> [19] terra_1.9-11 codetools_0.2-20 htmltools_0.5.9
#> [22] sass_0.4.10 yaml_2.3.12 pkgdown_2.2.0
#> [25] pillar_1.11.1 jquerylib_0.1.4 BiocParallel_1.46.0
#> [28] DelayedArray_0.38.1 cachem_1.1.0 magick_2.9.1
#> [31] abind_1.4-8 tidyselect_1.2.1 digest_0.6.39
#> [34] bookdown_0.46 labeling_0.4.3 fastmap_1.2.0
#> [37] grid_4.6.0 cli_3.6.6 SparseArray_1.12.2
#> [40] magrittr_2.0.5 S4Arrays_1.12.0 withr_3.0.2
#> [43] scales_1.4.0 rmarkdown_2.31 XVector_0.52.0
#> [46] ragg_1.5.2 beachmat_2.28.0 evaluate_1.0.5
#> [49] knitr_1.51 viridisLite_0.4.3 rlang_1.2.0
#> [52] Rcpp_1.1.1-1.1 glue_1.8.1 scuttle_1.22.0
#> [55] BiocManager_1.30.27 jsonlite_2.0.0 R6_2.6.1
#> [58] systemfonts_1.3.2 fs_2.1.0